





Introduction
Pancreatic cancer is not failing because researchers lack targets. The field has identified KRAS, its effectors, its microenvironmental enablers, and its metabolic dependencies with considerable precision over the past two decades. The failure is not a shortage of biological knowledge. It is a mismatch between the level at which biology is understood and the level at which therapeutic strategy is designed.
Most drug programs in pancreatic ductal adenocarcinoma still operate by selecting a node and blocking it. The logic is intuitive. Identify a protein that cancer cells depend on, disrupt its function, and watch the tumor regress. This model works with satisfying regularity in vitro and collapses with equal regularity in vivo. Understanding why requires stepping back from individual targets and looking at the networked, adaptive, metabolically constrained system that KRAS-mutant PDAC actually is.
The story of CCL-114 and CCL-115 is the story of that shift in understanding, forced by data that could not be explained by the original hypothesis.
When Early Data Revealed More Than Expected
The CCL-114 and CCL-115 series originated from cannabinoid-inspired chemistry, approached with a framework built around receptor engagement, reactive oxygen species activity, and pathway-level target interaction. The early data were encouraging in the ways that early oncology data typically are. Both compounds showed strong in vitro potency against PDAC cell lines, meaningful activity in three-dimensional tumor spheroid systems, and tumor growth inhibition in initial in vivo studies.
The original framing was that receptor engagement drove the effect. This is a reasonable hypothesis when the chemistry has cannabinoid structural inspiration and ROS activity is measurable. What made these compounds scientifically interesting was not that they worked under this framework. It was that they kept working when the framework should have predicted failure.
Resistant PDAC models, which express the bypass machinery that defeats pathway-specific inhibitors, showed sensitivity. Heterogeneous tumor environments, which create selective pressure that typically erodes target-dependent activity, did not eliminate the compounds’ efficacy. The conditions under which single-pathway drugs routinely fail were not conditions under which CCL-114 and CCL-115 failed.
A Forced Rethinking of the Target
Data that exceeds the explanatory capacity of its hypothesis is, in oncology, a rare and valuable signal. When compounds retain activity in resistant environments where their supposed mechanism of action should be neutralized, one of two things is true: either the resistance data is wrong, or the mechanistic hypothesis is incomplete.
In this case, the mechanistic hypothesis was incomplete. What the behavior of CCL-114 and CCL-115 across multiple model systems pointed toward was not a receptor or a pathway but a property of the cancer cell’s biochemical context that cuts across all survival branches simultaneously. That property is redox balance.
KRAS-mutant PDAC cells are chronically stressed oxidatively. Their antioxidant systems are already operating near capacity to manage a baseline ROS burden that far exceeds what normal cells experience. This is not incidental to the cancer phenotype. It is a consequence of the same metabolic reprogramming that makes these cells grow rapidly, consume glucose aggressively, and survive in nutrient-poor microenvironments. The elevated oxidative state that makes them metabolically competitive also makes them exquisitely vulnerable to compounds that perturb redox equilibrium rather than blocking discrete signaling events.
This is the insight that the CCL-114 and CCL-115 data forced. The question was no longer which node to block. It became how hard the redox balance needed to be perturbed before the system could no longer compensate.
Redox Perturbation and the PDX Model Evidence
Patient-derived xenograft models represent the most demanding preclinical test available for PDAC compounds. Unlike cell line xenografts derived from long-passaged laboratory strains, PDX tumors retain the genetic heterogeneity, stromal architecture, and adaptive signaling capacity of the human tumors from which they are derived. Compound activity in a PDX model means something fundamentally different from activity in a homogeneous cell line, because it has survived the biological complexity that most preclinical candidates cannot navigate.
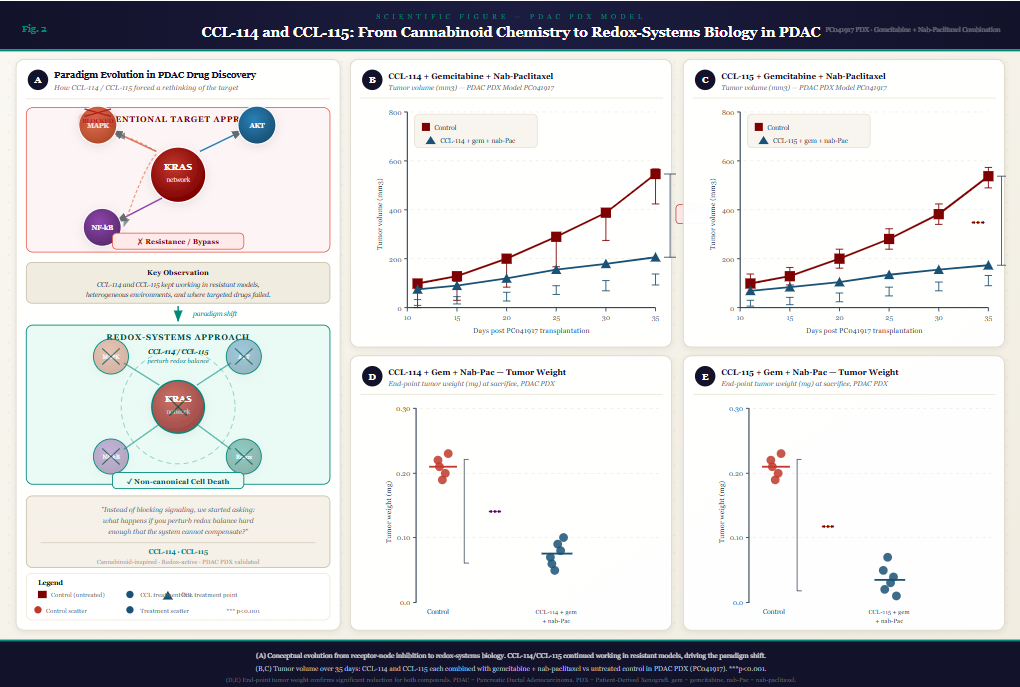
In the PC041917 PDX model, CCL-114 and CCL-115 were each evaluated in combination with gemcitabine and nab-paclitaxel, the current standard of care regimen in pancreatic cancer. Both compounds produced statistically significant reductions in tumor volume and tumor weight compared to untreated controls across the 35-day measurement period, with statistical significance reaching p less than 0.001 in all four readouts. Tumor weight at sacrifice was dramatically lower in both treatment groups, with CCL-115 producing particularly pronounced reduction.
These results are significant not only because of the magnitude of the effect, but because of the biological context in which they were achieved. PDAC PDX tumors are not cooperative targets. They carry the same redundant signaling architecture, stromal shielding, and adaptive capacity that causes resistance in the clinic. An agent that produces significant growth inhibition in this setting, combined with standard of care rather than as a single agent, is demonstrating that its mechanism survives biological complexity.
What Redox Systems Thinking Changes About Drug Design
The conceptual shift represented by CCL-114 and CCL-115 has implications that extend beyond these two compounds. Most PDAC drug programs are built around the assumption that the disease has a targetable weak point: one node in the network that is sufficiently essential that its loss cannot be compensated. The clinical record of the past fifteen years suggests this assumption is wrong.
KRAS-mutant PDAC does not have a single essential node in the sense that conventional drug design requires. It has a survival network with multiple redundant inputs, and the more precisely a drug is designed to hit one of them, the more efficiently the network adapts around the intervention. Resistance is not a failure of drug potency. It is a predictable output of a system that evolved under exactly this kind of selective pressure.
Targeting redox balance rather than signaling nodes addresses this problem at its source. Perturbing the oxidative environment that all branches of the KRAS survival network depend on simultaneously denies the system the adaptive options that defeat pathway-specific inhibitors. It is not a more potent intervention. It is an intervention designed at the correct level of biological organization.
Conclusion
CCL-114 and CCL-115 began as cannabinoid-inspired compounds with a receptor-based mechanistic hypothesis. They became evidence for a different way of thinking about PDAC, driven by the observation that they continued working in conditions where their original framing predicted they would not. The PDX data from the PC041917 model, with statistically significant tumor volume and weight reductions across both compounds in combination with standard of care, confirms that the redox-systems approach survives the most demanding preclinical standard available.
Pancreatic cancer does not need more targets. It needs a different level of biology. The work on CCL-114 and CCL-115 is an early but substantive indication that perturbing the redox context of a networked survival system may be the kind of intervention that can meet that standard.