




Introduction
For decades, pancreatic ductal adenocarcinoma has been described in shorthand as a KRAS-driven disease. This framing is scientifically accurate in the narrowest sense, KRAS mutations are present in approximately 95 percent of PDAC cases, making it one of the most mutation-consistent cancers in human oncology. But accuracy at the level of mutation frequency does not translate into completeness at the level of biological understanding. Describing PDAC as KRAS-driven is a bit like describing a city’s infrastructure problem as a traffic light malfunction. The traffic light may be broken, but the congestion it causes ripples across an entire interconnected road system.
This distinction matters enormously for therapeutic design. Most oncology programs targeting PDAC have, for years, focused on shutting down KRAS or one of its immediate downstream effectors. And most of them have failed, not in the laboratory, but at the level of clinical translation. Understanding why requires a deeper examination of how KRAS actually operates within the cancer cell, not as an isolated switch, but as the master regulator of a complex and adaptive survival network.
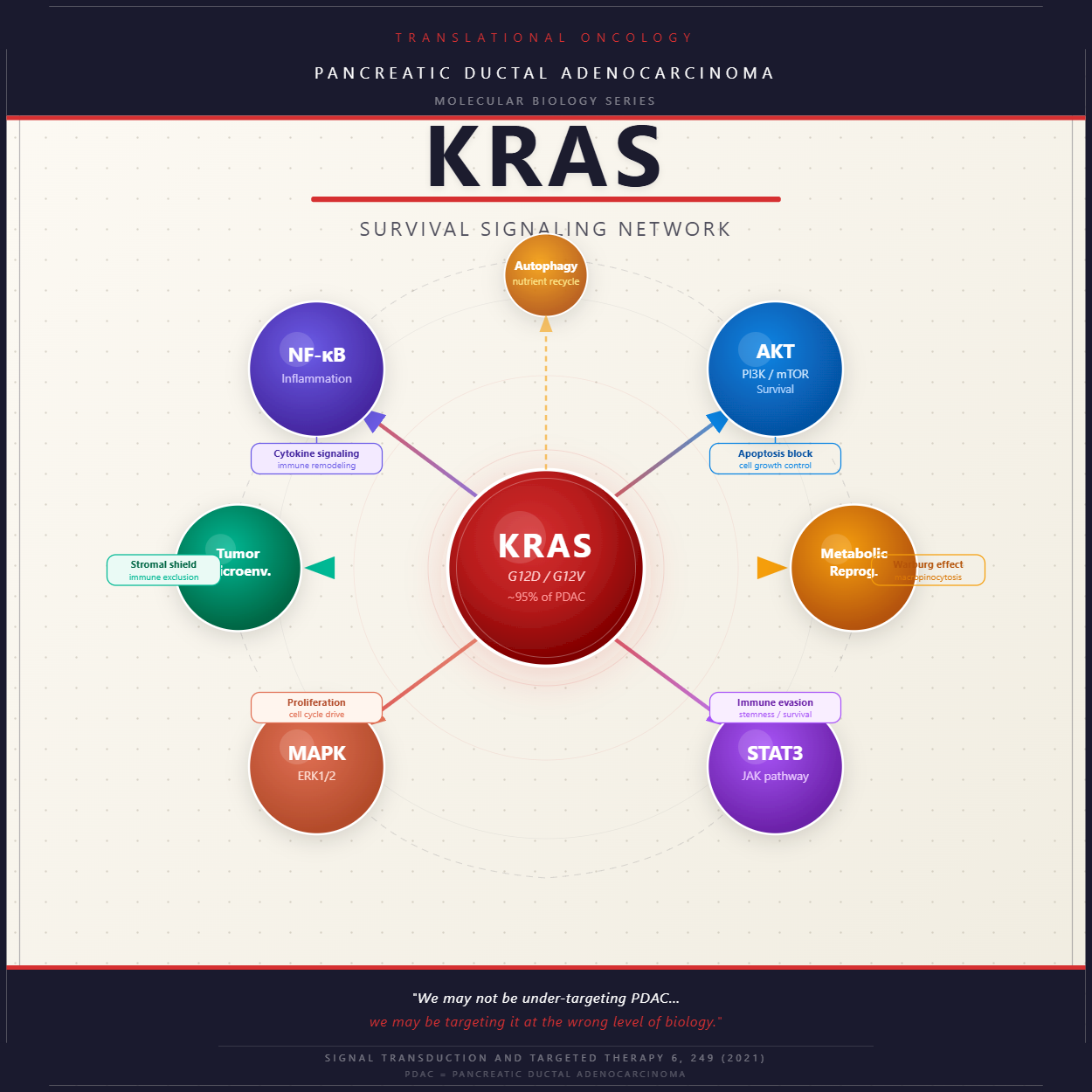
KRAS Does Not Act Alone – It Orchestrates a Multi-Pathway Survival Architecture
The biological reality of KRAS signaling in PDAC is far more complex than a single oncogene driving tumor growth. When KRAS is mutated and constitutively active, it simultaneously activates and sustains multiple downstream pathways that collectively ensure the cancer cell’s survival, proliferation, and resistance to therapeutic intervention.
The MAPK/ERK cascade is the most widely recognized arm of this network. Activated by mutant KRAS through RAF and MEK, this pathway drives relentless cellular proliferation and suppresses the normal checkpoints that would otherwise halt uncontrolled division. Running in parallel, the PI3K/AKT/mTOR axis provides a second layer of survival signaling, blocking apoptosis and coordinating cellular growth with nutrient availability.
Beyond these two canonical branches, mutant KRAS activates inflammatory programs through NF-κB and STAT3. These transcription factors are not merely passengers in PDAC biology – they actively remodel the tumor microenvironment, promote immune evasion, and sustain a chronic inflammatory state that accelerates tumor progression. STAT3 in particular has been linked to cancer stem cell maintenance, meaning its activity contributes not only to tumor growth but to the reservoir of cells capable of regenerating the tumor after treatment.
Finally, KRAS-mutant PDAC cells rely heavily on metabolic reprogramming to survive in the nutrient-poor environment of the pancreatic stroma. This includes the Warburg effect, in which glucose is preferentially metabolized through glycolysis even in the presence of oxygen, as well as macropinocytosis, a process by which cancer cells engulf extracellular material to scavenge amino acids and lipids. Autophagy – the cellular self-digestion pathway – adds yet another metabolic escape route, allowing cancer cells to cannibalize their own components for energy under conditions of stress.
Taken together, these pathways do not operate independently. They are interconnected through extensive cross-talk, forming a resilient network in which the activity of one branch can compensate for suppression of another.
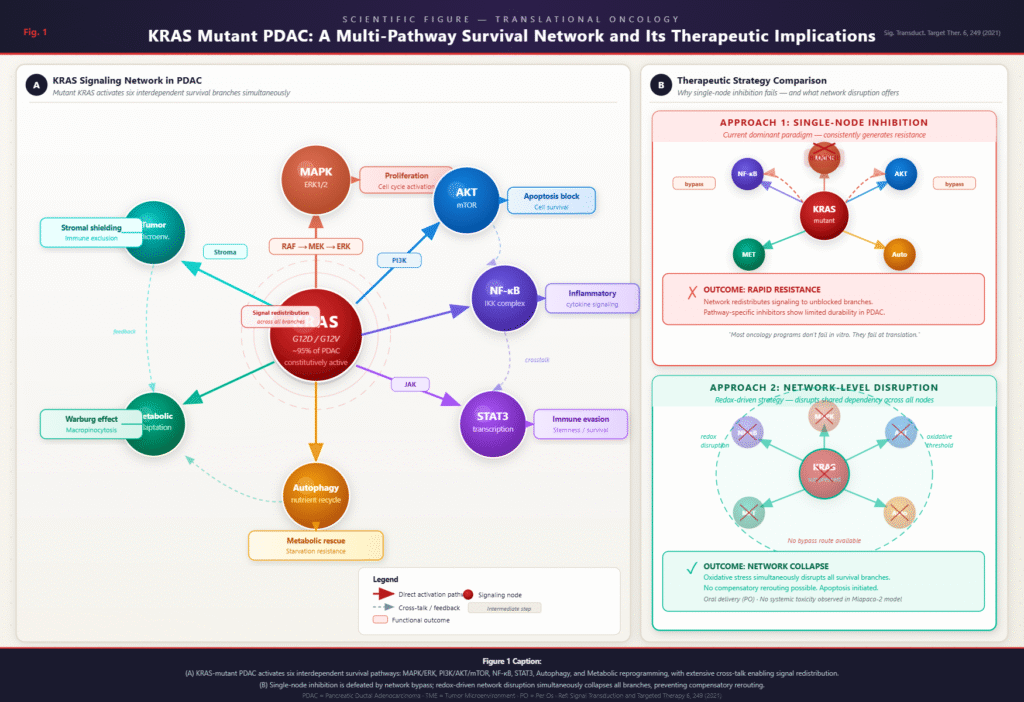
Why Single-Node Inhibition Consistently Generates Resistance
The logical implication of a networked survival architecture is that any therapeutic strategy targeting only one component of that network will eventually fail. This is not a theoretical prediction – it is a well-documented empirical pattern that has repeated itself across PDAC drug development programs.
When a pathway-specific inhibitor suppresses MAPK/ERK signaling, for example, KRAS-mutant cells do not simply die. They upregulate PI3K/AKT activity, reroute survival signaling through NF-κB, or increase autophagic flux to compensate for the metabolic disruption. The network, having evolved under constant selective pressure, redistributes its activity to maintain the critical functions the inhibited node was providing. This is not random biological noise. It is a highly coordinated adaptive response.
The clinical history of MEK inhibitors in PDAC reflects this phenomenon precisely. Despite strong preclinical rationale and measurable target inhibition in early studies, MEK inhibitors have consistently failed to produce durable responses in patients. The tumors respond briefly, then regrow through alternative pathway activation. The same pattern has emerged with attempts to directly target KRAS G12D and G12V, where initial responses have been followed by rapid resistance emergence through bypass mechanisms.
This failure mode points to a fundamental problem with the prevailing therapeutic paradigm. If the goal is to eliminate a cancer cell that depends on multiple interconnected survival mechanisms, then sequentially blocking individual components of that network is unlikely to achieve lasting disease control. Each inhibition event creates selective pressure that drives the evolution of resistance through the very pathways left untouched.
The Case for Network-Disruption Strategies in PDAC
If the limitation of single-node inhibition is the network’s ability to reroute signaling, then the logical therapeutic response is to disrupt the network at a level that does not permit easy rerouting. This is the conceptual foundation that makes redox-based and other multi-target strategies scientifically compelling in PDAC.
Oxidative stress disruption, for instance, does not block one specific downstream effector of KRAS. Instead, it destabilizes the biochemical environment on which all downstream signaling depends. When reactive oxygen species are elevated beyond the cancer cell’s compensatory capacity, the scaffolding that supports the entire network becomes compromised simultaneously. NF-κB, STAT3, AKT, and ERK all require a stable redox environment to function. A strategy that targets that shared dependency is inherently more resistant to the bypass mechanisms that defeat single-node inhibitors.
This does not mean that network disruption is simple to achieve or without risk. The challenge is selectivity – ensuring that the disruption preferentially affects cancer cells rather than normal tissue. But the scientific rationale for pursuing this class of strategy in PDAC is grounded in a clear-eyed understanding of what has failed before and why. Addressing PDAC at the wrong level of biology, no matter how precisely executed, will continue to generate the same pattern of transient response and rapid resistance that has defined this disease’s therapeutic history.
The field may not be under-investing in PDAC research. It may be investing in the wrong conceptual framework – one that treats a dynamic, adaptive network as if it were a simple on-off switch. Recognizing that distinction is the first step toward designing interventions that operate at the right level of biological complexity.
Conclusion
KRAS is not a single therapeutic target. It is the central node of a multi-layered signaling network that pancreatic cancer cells have evolved to depend on in its entirety. Pathway-specific inhibitors fail not because they are poorly designed, but because the network responds to their pressure by redistributing survival signaling through alternative routes. Durable therapeutic progress in PDAC will require strategies capable of disrupting that network at a systems level, rather than attempting to defeat it one node at a time. The biology of this disease demands a more ambitious – and more accurate, conceptual framework than single-gene targeting has ever been able to provide.