





Introduction
In oncology drug discovery, the point at which a compound’s mechanism stops fitting neatly into a known pathway is usually the point at which interest fades. Programs are built around mechanistic narratives. When the narrative breaks down, confidence in the program typically follows. What makes CCL-106 scientifically unusual is that the mechanism stopped making easy sense and the compound kept working anyway.
Cells continued to die. Tumors continued to shrink. Combination with chemotherapy produced better outcomes than either agent alone. The in vivo data held. What did not hold was the assumption that a clean pathway explanation was required before that data could be trusted. Understanding what this means for PDAC drug discovery requires stepping back from the reflex to categorize cell death and asking a more fundamental question about what the compound is actually doing to the cancer cell as a system.
The Problem with Pathway-Centric Mechanistic Framing
The standard approach to explaining why a compound kills cancer cells is to assign it to one of the recognized cell death modalities. Apoptosis is the most familiar, a regulated dismantling of the cell through caspase cascades that can be blocked experimentally to test whether it is responsible. Ferroptosis, an iron-dependent form of oxidative cell death, has attracted significant attention in cancer biology because of its connection to lipid peroxidation and redox dysregulation. Autophagy, the cellular self-digestion program, can under some conditions promote rather than prevent death and is therefore also interrogated in mechanistic studies.
With CCL-106, each of these pathways was examined. Blocking apoptosis did not rescue the cells. Ferroptosis markers were present but incomplete, suggesting partial involvement without full explanatory power. Inhibiting autophagy did not alter the outcome. Taken individually, none of these findings is unusual. What is unusual is the combination: a compound that produces reproducible, significant cell death in vitro and measurable tumor reduction in vivo, combined with chemotherapy enhancement, while resisting assignment to any single canonical death program.
The temptation in this situation is to keep searching for the missing pathway. The more productive response is to question whether a pathway is the right unit of analysis at all.
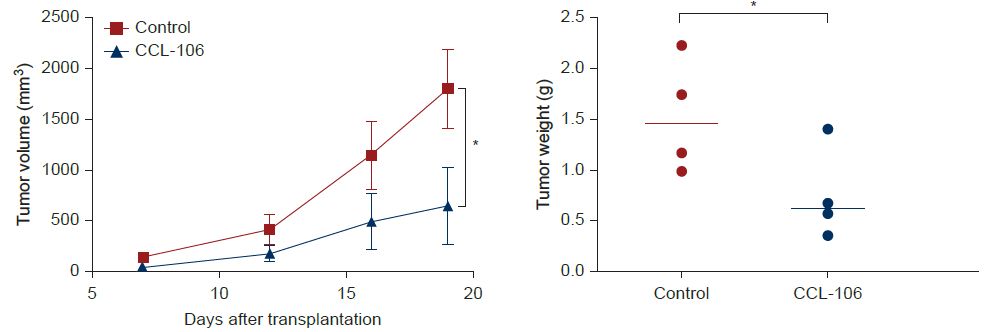
The Redox Metabolic Breaking Point Hypothesis
PDAC cells exist in a state of chronic oxidative stress. The same metabolic reprogramming that drives their aggressive growth, their resistance to nutrient deprivation, and their tolerance of the hypoxic tumor microenvironment also forces them to operate near the upper limit of their antioxidant capacity. Their redox homeostasis is not comfortably maintained. It is actively managed, and that active management consumes resources that would otherwise support adaptive responses to therapeutic stress.
The hypothesis that CCL-106 is pushing cells past a redox and metabolic breaking point rather than activating a specific death pathway offers a coherent explanation for the observed data. If the relevant event is not the engagement of a particular death program but the creation of conditions from which the cell’s recovery machinery cannot restore equilibrium, then the absence of a clean pathway signature is expected rather than puzzling.
Apoptosis, ferroptosis, and autophagy are not mutually exclusive with this model. They may all be downstream responses to a collapse in redox and metabolic integrity, each occurring to a partial degree because the initiating event is upstream of the branching point where these programs diverge. The cell is not dying by apoptosis. It is dying because it cannot recover, and the fragmented markers of multiple death pathways reflect the biological disorder of that collapse rather than the engagement of any one of them.

Why Chemotherapy Enhancement Changes the Picture
The observation that chemotherapy works better in combination with CCL-106 is the most clinically significant finding in the current data and the one that most directly supports the breaking point hypothesis. Chemotherapeutic agents such as gemcitabine and nab-paclitaxel impose metabolic and genotoxic stress on cancer cells. PDAC cells resist this stress through the same adaptive machinery that allows them to survive in hostile microenvironments: upregulation of DNA damage response, metabolic flexibility, and antioxidant buffer capacity.
If CCL-106 is already compressing that adaptive capacity through redox perturbation, then the residual buffer available to resist chemotherapy is reduced. The combination does not simply add two toxic insults. It sequentially depletes the adaptive resources that chemotherapy would otherwise encounter intact. The cancer cell cannot compensate for both challenges simultaneously because the resource pool that would enable compensation has already been destabilized.
This is a mechanistically coherent basis for combination synergy that does not require a precise pathway-level explanation. It requires only that the two agents impose stress on overlapping adaptive systems, which is consistent with everything observed in the CCL-106 data to date.
Conclusion
CCL-106 is at an early stage of mechanistic understanding, and that early stage is precisely where the most important scientific questions are still open. The in vivo tumor reduction is real and statistically significant. The chemotherapy synergy is real. The absence of a clean canonical death pathway explanation is real. The most productive interpretation of this combination of findings is not that the mechanism is elusive, but that it is operating at a level of biological organization that pathway-centric analysis is not designed to capture.
The question worth pursuing is not which pathway CCL-106 is hitting. It is what happens to a PDAC cell when its capacity to recover from redox and metabolic disruption is exhausted. If that capacity is the actual vulnerability being exploited, then the behavior of CCL-106 across resistant models, in combination with chemotherapy, and without a clear pathway signature is exactly what the hypothesis predicts. That coherence is where the science gets genuinely interesting