




Predictive Docking Strategies: Leveraging Protein Pocket Analysis for Lead Optimization
In the highly competitive field of pharmaceutical research, the ability to accurately predict how a small molecule interacts with its biological target is a cornerstone of efficient drug design. While ligand-based Structure-Activity Relationship (SAR) modeling has traditionally provided a foundation for understanding potency, the integration of structure-based hypotheses offers a more granular view of molecular behavior. By utilizing advanced computational platforms like StarDrop, researchers can now move beyond two-dimensional data to visualize the spatial orientation of a compound within a protein’s binding pocket. This shift toward structure-aware modeling allows for a more rational approach to scaffold fine-tuning, significantly reducing the time required to optimize binding affinities.
The Integration of Pocket Based Hypotheses in SAR Analysis
Traditional drug discovery often relies heavily on ligand-based SAR, where researchers analyze the properties of known active compounds to infer the requirements for potency. However, this method can be limited by a lack of structural context regarding the protein environment. By incorporating the protein pocket into the analysis, scientists can generate a binding hypothesis that accounts for the physical constraints and electronic environment of the target site.
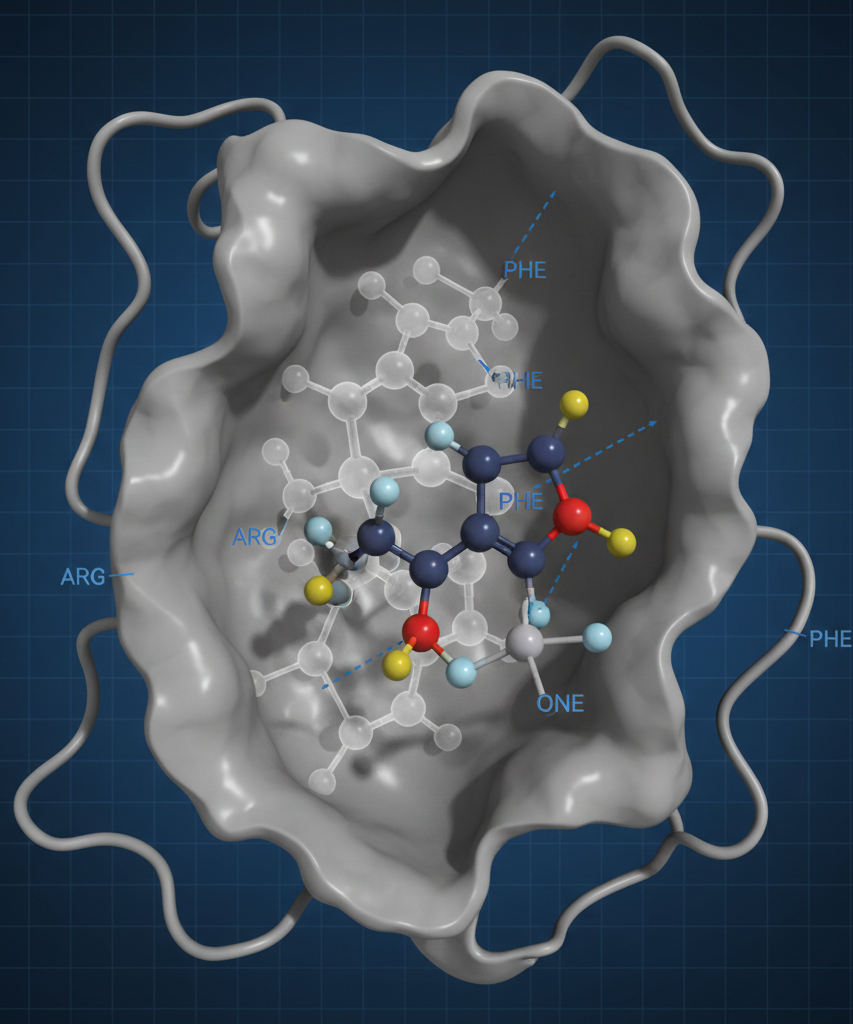
Using specific protein-ligand structures, such as the 4ACC crystal structure, serves as a benchmark for comparing novel compounds of interest. This comparison allows researchers to align their proprietary scaffolds with the original ligand, providing a direct visual of the three-dimensional space occupied by the reference molecule. Understanding these spatial relationships is vital for determining which functional groups are essential for maintaining contact with key amino acid residues and which areas of the scaffold can be modified to improve pharmacokinetic properties without disrupting the binding pose.
Fine Tuning Scaffolds through 3D Structural Alignment
The process of scaffold optimization is a delicate balance of maintaining inhibitory activity while improving drug-like properties. Through the use of 3D structural alignment, medicinal chemists can see exactly how a proposed compound positions itself within the protein pocket relative to a validated lead. This visual data provides a rationale for why certain binding affinities are high, as researchers can observe the overlapping volumes and specific interactions, such as hydrogen bonding or hydrophobic packing, that drive the association.
Fine-tuning a scaffold based on these alignments allows for the “de-risking” of chemical synthesis. Instead of synthesizing dozens of analogs to probe a pocket, researchers can focus on the most promising orientations. This predictive docking approach helps in identifying “clashes” where a molecule might be too bulky for a specific sub-pocket or “voids” where a molecule could be extended to pick up additional binding energy. By seeing the 3D space of the original ligand, the design process becomes an exercise in precision engineering rather than trial and error.
Implications for Binding Affinity and Lead Discovery
The primary goal of utilizing binding hypotheses is to achieve superior binding affinity through informed molecular design. High binding affinity is not just a measure of strength; it is a prerequisite for reducing the therapeutic dose and minimizing off-target effects. When computational models successfully predict the positioning of a compound, the resulting data helps explain the underlying thermodynamics of the binding event.
So far, the application of these structure-based insights has proven instrumental in explaining why certain chemical series exhibit high potency. It provides a bridge between raw experimental data and the physical reality of molecular recognition. For industry professionals, these insights are crucial for securing patent protection and moving a lead series toward the preclinical stage. As computational tools become more sophisticated, the ability to decode the “dance” between ligand and protein pocket will remain the gold standard for innovative drug development.
Conclusion
The evolution of drug discovery continues to be driven by the synergy between experimental chemistry and computational modeling. By leveraging protein pocket analysis and generating high-fidelity binding hypotheses, researchers can navigate the complex landscape of lead optimization with greater confidence. Tools that allow for the alignment of new compounds against established crystal structures provide the structural clarity needed to refine chemical scaffolds and maximize binding affinity. As we continue to integrate these 3D insights into the standard SAR workflow, the path from initial hit to optimized lead becomes shorter, more predictable, and ultimately more successful in delivering effective therapeutics