




Introduction
There is a recurring pattern in pancreatic cancer drug development that clinical researchers and translational scientists know well but rarely name directly. A compound shows strong activity in vitro. It engages its target. Preclinical data looks promising. Then it enters a real tumor system, and the tumor does something unexpected. Not dramatic failure, not complete insensitivity, but a kind of biological negotiation in which the tumor absorbs the therapeutic pressure, shifts its internal organization, and continues to grow.
This is not a pharmacological problem in the conventional sense. The drug is reaching its target. The target is being inhibited. The downstream pathway is being disrupted. And yet the overall outcome remains inadequate. The reason is that the tumor has not simply been disrupted. It has adapted. And in PDAC, that adaptation happens faster and more completely than almost any drug program is designed to account for.
Understanding this pattern, and what it demands of therapeutic strategy, is the most important conceptual problem in PDAC drug development today.
The Adaptation Cycle That Defeats Most Drugs
The tumor’s adaptive response to therapeutic pressure follows a recognizable cycle that repeats across different drug classes and different target combinations. Each step in the cycle represents a different layer of adaptive capacity that the tumor can deploy in response to a specific type of intervention.
When a pathway is inhibited, the tumor reroutes. KRAS-mutant PDAC maintains multiple redundant signaling branches that can compensate for the loss of activity in any single pathway. Block MAPK, and AKT signaling increases. Suppress PI3K, and NF-kB activity compensates. The network is not organized around any single node. It is organized around resilience, and resilience means that losing one input does not collapse the system.
When the dose is increased to overcome partial resistance, the tumor compensates. The antioxidant and stress response systems that keep the cancer cell viable under conditions of elevated therapeutic stress are upregulated. Metabolic flexibility, already a defining feature of PDAC biology, allows the cell to shift its energy metabolism in ways that buffer the impact of the higher drug exposure. The system does not simply absorb more damage. It reorganizes its resource allocation to tolerate it.
When therapies are combined to address multiple pathways simultaneously, the tumor reorganizes at a deeper level. Transcriptional programs that control survival gene expression are remodeled. Epigenetic states shift. The tumor draws on the stromal and immune components of its microenvironment for additional metabolic and signaling support. The combination produces a more complex adaptive response than any single agent, but the response still occurs, and it still allows the tumor to persist.
Why This Is a Systems Problem, Not a Target Problem
The conclusion that this adaptation cycle forces is uncomfortable for a field built around the discipline of target identification and drug design. If the tumor can reroute around any single inhibited node, compensate for any escalation in dose, and reorganize around any combination of targeted agents, then the problem is not which target to choose. It is whether target-based thinking is the appropriate framework at all.
The tumor is not responding to individual drug-target interactions in isolation. It is responding as an integrated system to the overall perturbation the therapy creates. The system has inputs, outputs, feedback mechanisms, and redundancy built into its architecture. It is organized, in evolutionary terms, to survive pressure, because the same selective forces that drove the cancer’s development also drove the development of its adaptive capacity.
A truly effective intervention in this context would need to impose perturbation at a level that the system’s adaptive mechanisms cannot address through rerouting, compensation, or reorganization. This means engaging not a specific node but a shared dependency that spans the entire system, something so fundamental to the cancer cell’s ability to function that no alternative pathway or metabolic shift can compensate for its disruption.
What CCL-106 Demonstrates in MIA PaCa-2
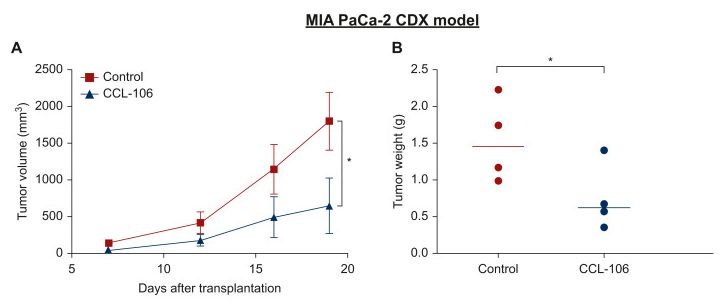
The MIA PaCa-2 CDX model represents one of the more demanding preclinical contexts in which to evaluate a PDAC compound. MIA PaCa-2 is an established pancreatic cancer cell line with well-characterized resistance properties and the kind of robust adaptive signaling architecture that makes it a meaningful proxy for the clinical challenge of PDAC. Results in this model carry more translational weight than results in more sensitive or cooperative cell systems.
CCL-106 administered orally in this model produced approximately 50 percent reduction in tumor volume compared to untreated control, with statistically significant reduction also observed in tumor weight at endpoint. The significance of these results is not only the magnitude of the reduction. It is that the reduction was achieved in a model where the tumor’s adaptive machinery is intact and active.
The working hypothesis for how CCL-106 achieves this involves redox perturbation at a level that exceeds the cancer cell’s compensatory capacity. Rather than blocking a specific signaling node that the network can reroute around, the approach imposes oxidative stress at a magnitude and breadth that the system’s antioxidant defenses, metabolic flexibility, and transcriptional stress responses cannot collectively manage. The adaptive cycle that defeats most drugs is broken not by finding a better node to block, but by shifting the level of intervention to a shared biochemical dependency that the node-blocking strategy leaves entirely intact.

Conclusion
PDAC tumors do not fail to respond to drugs because the targets are wrong or the compounds are too weak. They fail to respond durably because the tumor is a system, and the system adapts faster than any target-based intervention can neutralize the vulnerability it exploits. Rerouting, compensation, and reorganization are not failures of drug resistance in the classical sense. They are the baseline operating behavior of a highly adaptive biological network under therapeutic pressure. The programs that will eventually produce durable benefit in PDAC are those designed to engage with the system rather than a node within it, to impose perturbation at a level that the adaptive machinery cannot negotiate around, and to do so in real tumor models where the system’s full adaptive capacity is present from the beginning.