




The Evolution of Structure Activity Relationship Modeling
In the field of medicinal chemistry, the process of developing a lead compound into a viable therapeutic candidate has traditionally been a labor intensive endeavor. Structure Activity Relationship (SAR) analysis is the cornerstone of this process, requiring researchers to systematically modify chemical scaffolds to observe changes in biological activity. Historically, this meant manual design cycles where chemists would brainstorm dozens or hundreds of analogs, a task that could consume weeks of specialized effort. However, the integration of advanced computational plugins, such as the Nova module within the StarDrop platform, is fundamentally shifting this paradigm. By automating the generation and evaluation of molecular analogs, researchers are now capable of exploring a vast chemical space with unprecedented speed and precision.
Accelerating Analog Generation Through Algorithmic Design
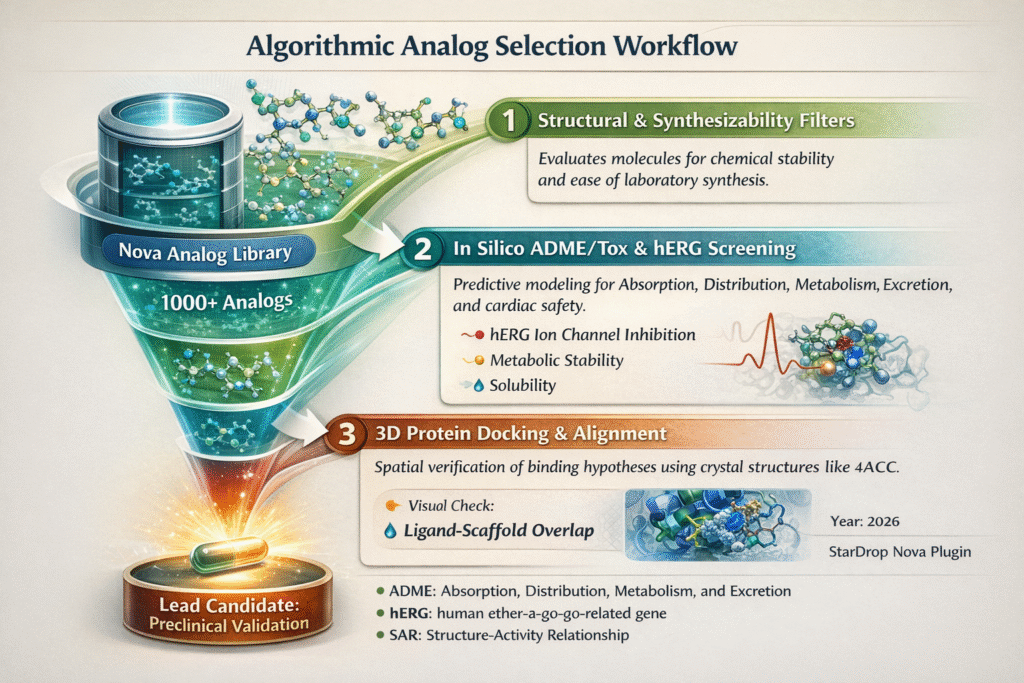
One of the most significant bottlenecks in early stage drug discovery is the conceptualization of chemical variations that maintain potency while improving safety profiles. Traditional methods often limit a researcher to a small number of derivatives based on personal experience or established literature. Automated design tools like the Nova plugin bypass these human limitations by applying diverse chemical transformation rules to a lead scaffold. In a fraction of the time it would take to manually design one hundred analogs, these systems can generate over one thousand unique molecular structures. This rapid expansion of the lead series allows for a more comprehensive investigation of the chemical landscape, identifying novel modifications that a computational novice might otherwise overlook.
Integrating Predictive Binding Affinity and Safety Profiles
The true power of modern computational SAR tools lies not just in their ability to generate structures, but in their capacity to evaluate them instantaneously. Generating a thousand analogs is of limited value if each must be manually screened; however, integrated platforms provide immediate feedback on binding information and predictive toxicology. For instance, when analyzing novel compounds for potential therapeutic use, the system can cross reference generated analogs against critical safety parameters, such as the hERG ion channel pathway, to flag potential cardiotoxicity risks early in the design phase.
By delivering binding information for a massive library of compounds in under two hours, these tools allow scientists to pivot from design to selection almost immediately. This rapid feedback loop is essential for identifying the most promising candidates for synthesis and subsequent in vitro experimental validation. The efficiency gained here represents a significant competitive advantage in the race to bring safe and effective therapeutics to market.
Bridging the Gap Between Computational Expertise and Laboratory Execution
The democratization of high level computational tools is transforming the role of the modern scientist. While the transition from manual design to automated SAR analysis involves a learning curve, the rapid results provided by these platforms accelerate the learning process itself. As researchers become more adept at utilizing algorithmic design, they can focus more on high level strategic decisions and the interpretation of complex data rather than the rote task of drawing chemical structures. This synergy between human insight and machine efficiency is the future of pharmaceutical research, ensuring that only the most robust and safe chemical entities proceed to preclinical and clinical animal studies.