
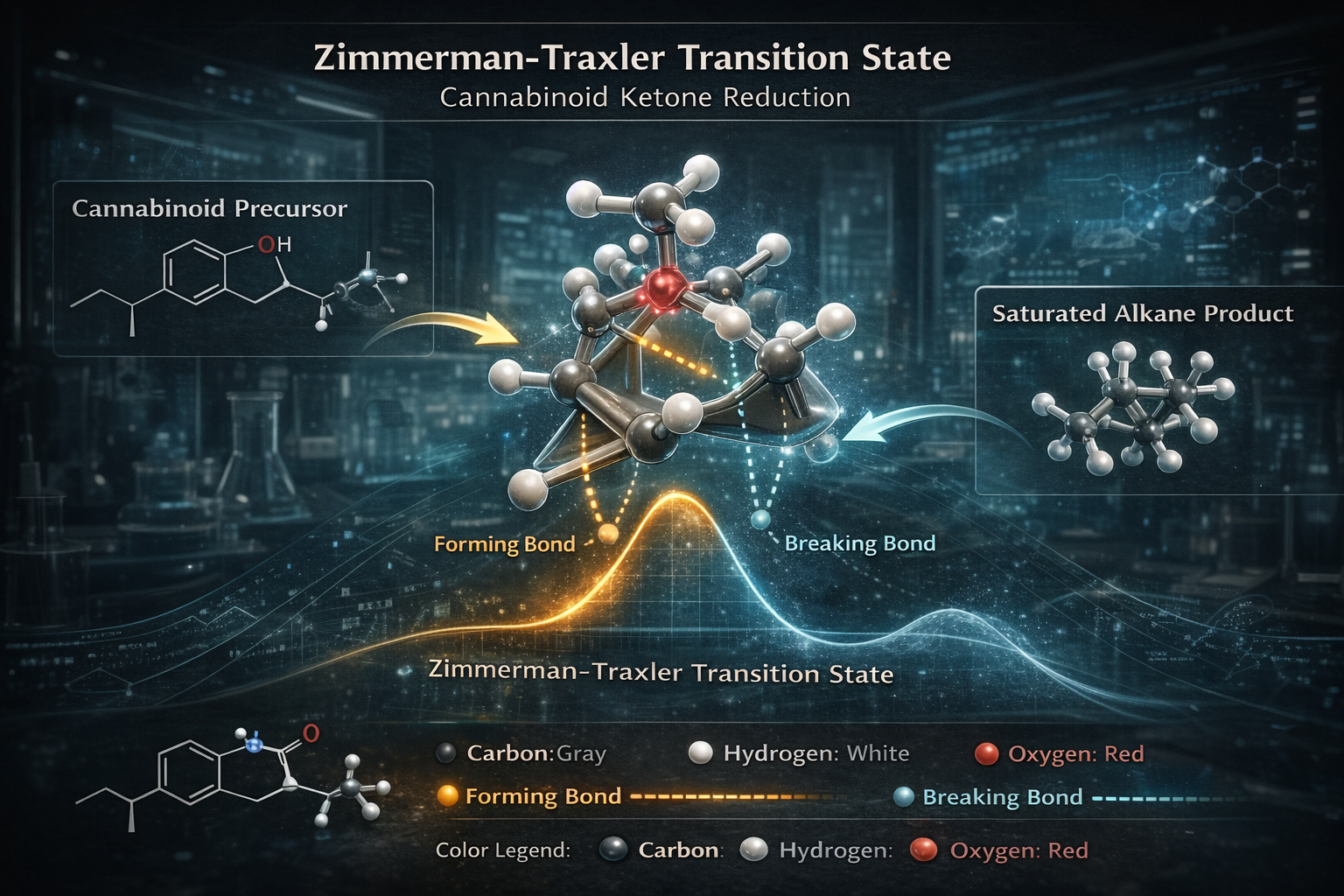




Predicting Diastereoselectivity: The Role of Transition State Models in Cannabinoid Synthesis
In the sophisticated field of medicinal chemistry, the ability to predict and control the stereochemical outcome of a reaction is paramount for developing effective therapeutic agents. Hydrogenation, a fundamental transformation used to saturate carbon-carbon double bonds or reduce carbonyl groups, often introduces new chiral centers into a molecule. For complex scaffolds like cannabinoids, where pharmacological activity is strictly dependent on three dimensional orientation, understanding the transition state is the difference between a successful synthesis and a failed batch. By applying classical and modern stereochemical models, researchers can navigate the energetic barriers of a reaction to favor the production of a specific isomer. This analytical precision is essential for the reproducible production of pure cannabinoid derivatives in both research and industrial settings.
Fundamental Models for Diastereoselective Carbonyl Reduction
The reduction of ketones and enones within a cannabinoid framework requires a deep understanding of steric and electronic influences. One of the most enduring tools for this analysis is the Zimmerman Traxler model. While often associated with aldol reactions, its principle of six membered chair like transition states is vital when hydrogenating alpha beta unsaturated ketones derived from cannabinoids. This model allows chemists to visualize the orientation of substituents, predicting which face of the molecule is more accessible to the reducing agent and thus which stereoisomer will predominate.
Complementing this is the Felkin Ahn model, which refined earlier rules by incorporating the importance of the staggered transition state and anti periplanar arrangements. In the selective reduction of cannabinoid ketones, Felkin Ahn analysis helps predict the major product by evaluating the steric bulk of groups adjacent to the carbonyl. By positioning the largest neighboring group perpendicular to the carbonyl pi system, researchers can determine the optimal trajectory for hydride attack or hydrogen addition. These models provide a theoretical framework that transforms complex molecular structures into predictable reaction paths, ensuring that the resulting cannabinoids possess the intended spatial configuration.
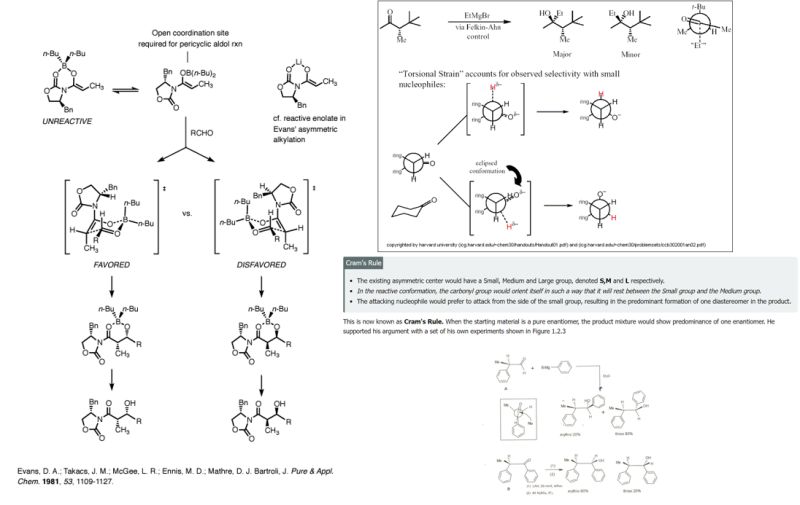
Chiral Induction and the Application of Cram’s Rule
When dealing with chiral intermediates in cannabinoid synthesis, the influence of existing stereocenters on newly formed ones is a critical consideration. Cram’s rule serves as a foundational technique for predicting the diastereoselectivity of additions to carbonyl groups attached to a chiral center. In the context of hydrogenation, this rule guides the selective addition of hydrogen by assessing the steric hindrance presented by small, medium, and large groups surrounding the active site.
As the cannabinoid industry moves toward enantioselective synthesis, understanding these models becomes even more important for chiral catalysis. The development of chiral catalysts for enantioselective hydrogenation often relies on the same principles of transition state stabilization found in the Zimmerman Traxler and Felkin Ahn models. By mastering these rules, scientists can design catalysts that mimic the biological specificity of enzymes, producing chiral cannabinoids with tailored pharmacological properties. This level of control is necessary for creating pharmaceutical grade products where the presence of an unwanted isomer could lead to off target effects or reduced efficacy.
Computational Simulation and Houk’s Model in Modern Research
As cannabinoid structures become increasingly complex, classical paper and pencil models are often supplemented with advanced computational methods. Houk’s model utilizes transition state theory in conjunction with modern density functional theory (DFT) to simulate the hydrogenation process. These computational simulations offer a detailed view of the transition states that are often too transient to observe experimentally.
By calculating the relative energies of competing transition states, researchers can gain insights into the potential products and product distributions with high precision. This is particularly useful for novel cannabinoids that do not fit neatly into traditional models. Computational analysis allows for the refinement of reaction parameters, such as temperature and solvent polarity, to enhance yield and selectivity before a single experiment is performed in the laboratory. This synergy between theoretical models and computational power is driving innovation in product development, allowing for the creation of unique cannabinoid derivatives with optimized stability and biological activity.
Conclusion
The integration of advanced transition state models like Zimmerman Traxler, Felkin Ahn, and Houk’s model is essential for pushing the boundaries of cannabinoid chemistry. These techniques allow researchers to move beyond trial and error, providing a rational basis for predicting reaction outcomes and optimizing synthesis. Accurate transition state analysis not only ensures the efficient production of desired isomers but also facilitates the design of novel cannabinoids with specific pharmacological profiles. As the industry continues to professionalize, the mastery of these organic chemistry principles will remain a defining factor in the development of the next generation of effective, high quality cannabinoid based products.