




Introduction
Kinase inhibitors remain one of the most important classes of small molecule therapeutics in modern drug discovery. Their ability to modulate signaling pathways makes them central to oncology inflammatory diseases and rare disorders. Identifying viable kinase inhibitor scaffolds requires a balance of chemical intuition structural insight and efficient computational workflows. As research teams grow more interdisciplinary accessibility of computational tools becomes just as important as their scientific power. Recent advances in user friendly in silico platforms are reshaping how teams explore binding interactions and optimize early stage chemical matter.
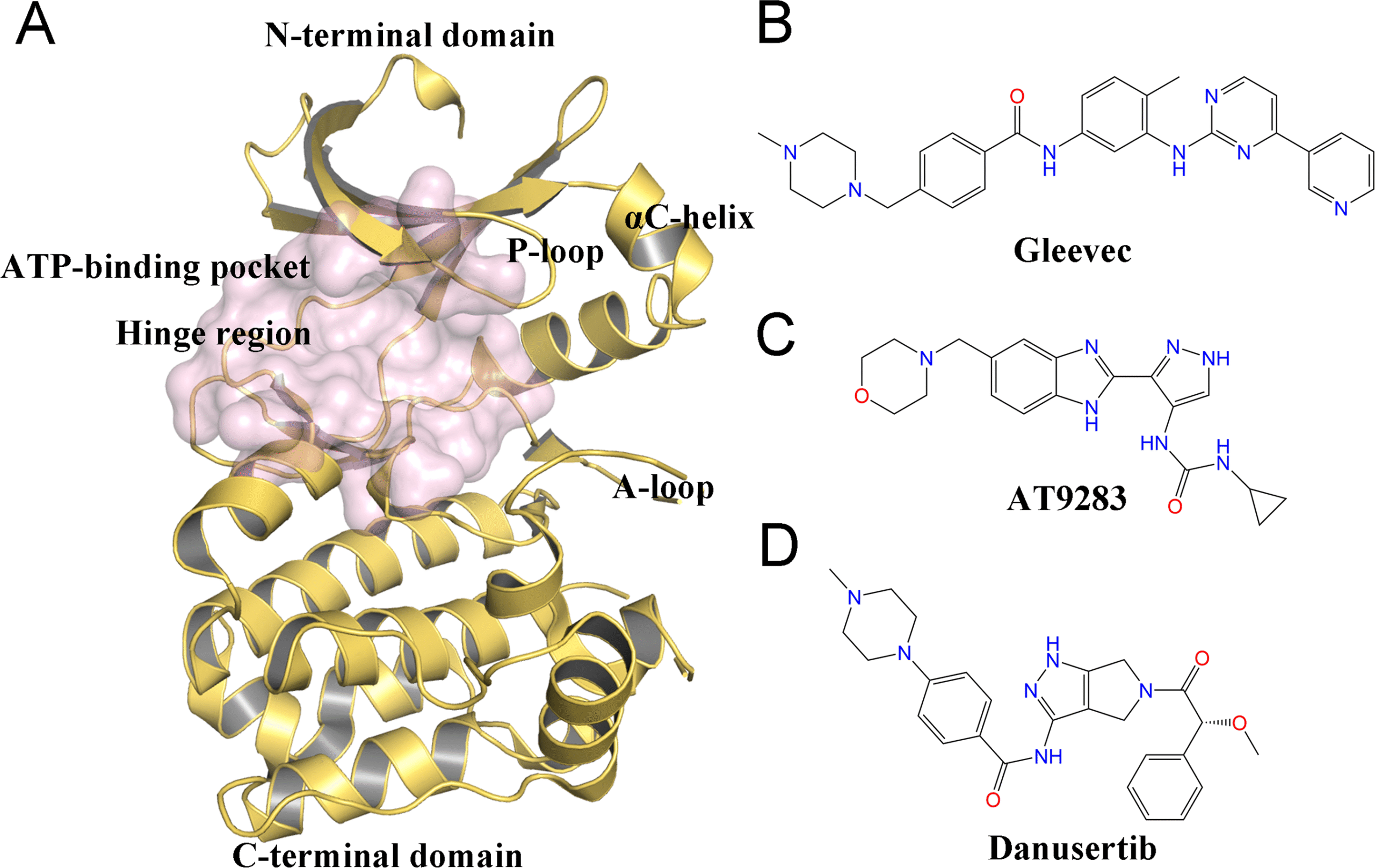
Identifying Promising Kinase Inhibitor Scaffolds
The first step in kinase focused discovery programs is scaffold identification. A scaffold must present functional groups capable of interacting with conserved regions of kinase active sites while retaining opportunities for selectivity. Computational screening allows researchers to evaluate shape complementarity hydrogen bonding potential and hydrophobic interactions before synthesis. This early filtering reduces experimental burden and focuses laboratory resources on the most promising chemical frameworks. When multiple scaffolds are evaluated in parallel computational insight becomes critical for prioritization.
Accessibility as a Catalyst for Computational Adoption
Historically powerful molecular modeling platforms often required specialized expertise limiting their use to a small number of trained individuals. While these tools delivered high quality predictions they also created bottlenecks within teams. Platforms such as StarDrop are changing this dynamic by emphasizing intuitive interfaces and streamlined workflows. Compared with more complex environments such as Schrodinger modern solutions allow scientists with diverse backgrounds to engage directly with binding analysis and structure activity exploration. This democratization of computational chemistry accelerates learning and encourages broader participation in molecular design.
Learning In Silico Binding Through Iterative Exploration
User friendly computational environments enable researchers to rapidly test hypotheses and visualize results without steep onboarding barriers. Exploring binding modes scoring trends and scaffold modifications becomes an interactive learning process rather than a gated technical task. Over time repeated exposure builds confidence and deepens understanding of molecular interactions. This hands on engagement strengthens the connection between computational predictions and experimental decision making ultimately improving the quality of candidate selection in kinase inhibitor programs.
Conclusion
The evolution of accessible in silico tools is transforming kinase inhibitor discovery. By lowering the barrier to entry for binding analysis and scaffold evaluation teams can move faster share knowledge more effectively and integrate computational insight earlier in research pipelines. As more scientists gain fluency in these methods the collective impact on drug discovery efficiency and innovation will continue to grow.