




Introduction
Computational chemistry has become an essential partner to experimental drug discovery. As molecular targets grow more complex and experimental timelines tighten researchers increasingly rely on in silico tools to guide decision making before synthesis begins. Structure based modeling allows scientists to visualize how compounds interact with proteins at an atomic level offering insight that traditional ligand only analysis cannot provide.
Modern computational platforms now integrate structure activity data with protein pocket information. This combination helps researchers move beyond correlation toward mechanistic understanding. By observing how molecules occupy three dimensional binding sites chemists gain clarity on why certain compounds perform better than others.
From Ligand Comparison to Binding Hypotheses
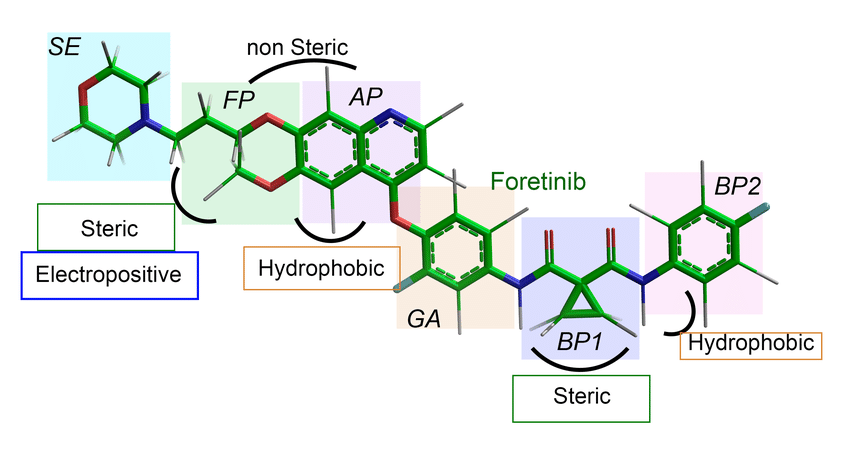
One of the most valuable aspects of structure guided computation is the ability to generate binding hypotheses. By aligning a known reference ligand within a protein pocket and comparing it to new compounds researchers can evaluate how closely novel scaffolds mimic key interactions.
This approach allows scientists to assess steric overlap orientation and functional group placement. Even small deviations in alignment can explain differences in affinity. Rather than relying solely on activity trends computational alignment provides a structural rationale for observed results.
Scaffold Refinement Through Protein Pocket Analysis
Protein pockets impose strict spatial constraints. Understanding these constraints enables precise scaffold refinement. Computational visualization reveals which regions of a compound interact favorably with the pocket and which regions introduce clashes or inefficiencies.
This insight supports rational modification of molecular frameworks. Adjustments can be made to improve complementarity with the binding site while preserving critical interactions. Such targeted refinement reduces unnecessary synthetic exploration and increases the probability of success.
Understanding High Binding Affinity Through Spatial Insight
High binding affinity is rarely accidental. It often arises from optimal positioning within the binding pocket combined with favorable intermolecular forces. Computational modeling makes these relationships visible.
By examining three dimensional occupancy researchers can identify why certain compounds outperform others despite similar chemical composition. This understanding informs future design cycles and strengthens confidence in experimental outcomes.
Conclusion
Structure based computational modeling transforms drug discovery from empirical iteration into informed exploration. By visualizing how compounds engage protein targets researchers gain predictive power that complements experimental data.
When computational insight and laboratory validation work together discovery becomes more efficient more explainable and more robust. Understanding molecular positioning within proteins is not just a technical advantage it is a strategic foundation for modern medicinal chemistry.