




Metabolic Architecture: Predictive Modeling of CYP450 Pathways in Drug Development
The pharmacological efficacy of any synthetic compound is fundamentally governed by its metabolic fate within the human body. As soon as a therapeutic agent enters systemic circulation, it is subjected to a complex array of enzymatic transformations designed to facilitate its clearance. The cytochrome P450 (CYP450) superfamily of enzymes represents the primary mechanism for the oxidative metabolism of drugs. For medicinal chemists, understanding the metabolic landscape of a scaffold is as critical as establishing its primary binding affinity. By leveraging in silico computational tools, researchers can now predict metabolic vulnerabilities early in the design phase, allowing for the strategic modification of labile sites to enhance the half life and stability of potential drug candidates.
The Role of P450 and Non P450 Enzymes in Molecular Degradation
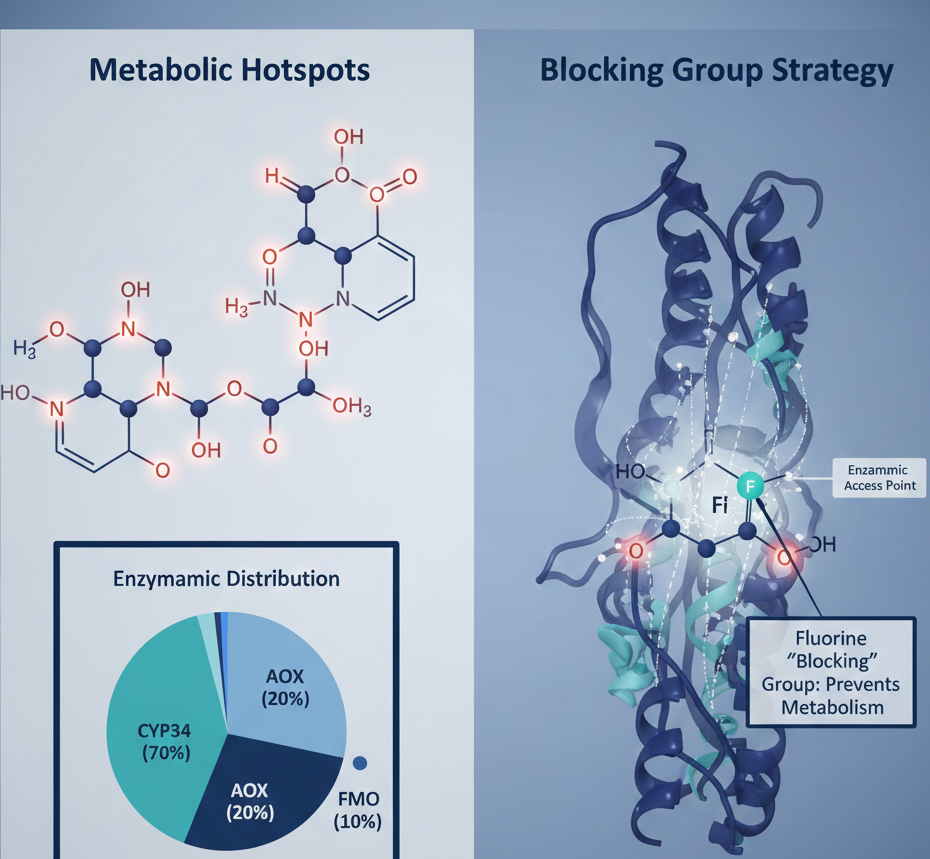
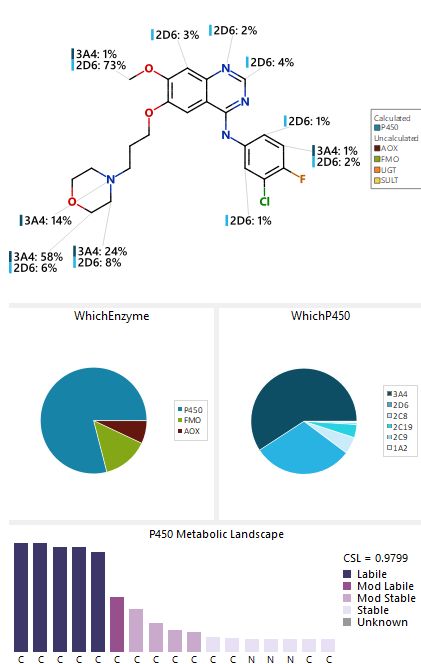
While CYP450 enzymes are the dominant force in phase I metabolism, they do not operate in isolation. Other enzyme systems, such as Aldehyde Oxidase (AOX) and Flavin containing Monooxygenase (FMO), also play pivotal roles in the oxidation and degradation of small molecules. These enzymes target specific functional groups, transforming lipid soluble compounds into more polar metabolites that the kidneys can easily excrete. For instance, the tyrosine kinase inhibitor Gefitinib exhibits a complex metabolic profile where multiple sites on the molecule are susceptible to enzymatic attack.
Predictive modeling allows scientists to visualize this metabolic landscape, identifying which atoms are most likely to undergo oxidation. This is often represented as a percentage of metabolism attributed to specific isoforms, such as CYP3A4 or CYP2D6. Understanding whether a molecule is labile, meaning easily metabolized, or stable is essential for optimizing the dose response relationship of a new chemical entity.
Strategic Structure Activity Relationship Modifications
Once a metabolic vulnerability is identified through in silico computation, the next step in the Structure Activity Relationship (SAR) workflow is to block these sites without compromising the compound’s biological activity. If a labile site is located in an area that is not critical for protein ligand interactions, chemists may introduce bioisosteres, such as substituting a hydrogen atom with a fluorine atom, to prevent enzymatic oxidation. This metabolic shielding can significantly increase the concentration of the drug in the blood over time.
However, a significant challenge arises when the site of metabolic attack coincides with a functional group necessary for binding to the target protein. In these cases, 3D molecular docking simulations become indispensable. Researchers must visualize the binding pocket to determine if a structural modification intended to prevent oxidation will create steric clashes within the protein environment. The goal is to design a molecule that fits perfectly within the pocket while simultaneously presenting a shielded profile to the body’s metabolic machinery.
Implications of Predictive Metabolic Profiling
The integration of metabolic prediction into the early design cycle has profound implications for the speed and cost of drug discovery. Traditional drug development often involved late stage failures where a highly potent compound was found to have a half life too short for clinical use. By identifying metabolic risks in silico, these failures can be avoided before a compound even reaches the laboratory bench.
Furthermore, predictive tools help in anticipating drug drug interactions. If a compound is primarily metabolized by a single isoform, such as CYP3A4, it may interact negatively with other medications that use the same pathway. Mapping the distribution of enzymes allows for the development of more robust therapeutic profiles that are safer for patients with complex medication regimens. This proactive approach to safety and efficacy is what separates modern precision medicine from historical trial and error methods.
Conclusion
The journey of a drug candidate from a computer model to a clinical success is defined by its ability to survive the body’s natural defense mechanisms. Catalytic enzymes like CYP450, AOX, and FMO are the gatekeepers of this process. By utilizing advanced computational tools to map the metabolic landscape of molecules like Gefitinib, medicinal chemists can make informed decisions that improve molecular stability and therapeutic potential. As our understanding of these enzymatic pathways grows, so too does our ability to engineer the next generation of medicines with optimized metabolic durability.